





SAÚDE
Presidente acompanha retomada da produção de insulina no Brasil em Nova Lima (MG)
Saiba mais sobre SAÚDE

PROTEÇÃO
Ministério da Saúde amplia vacinação da dengue para mais 6 estados
Saiba mais sobre PROTEÇÃO
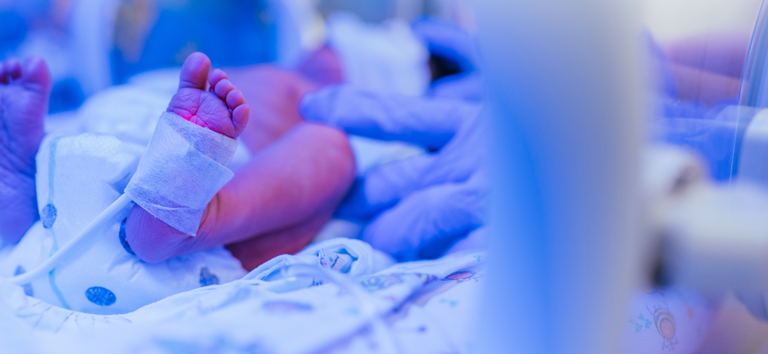
APOIO FINANCEIRO
Ministério da Saúde destina R$ 97 milhões para ampliar UTIs pediátricas
Saiba mais sobre APOIO FINANCEIRO

NOVO PAC SAÚDE
Publicada habilitação de 500 propostas de construção de novas UBS
Saiba mais sobre NOVO PAC SAÚDE



PROCESSO SELETIVO
Seleção de servidores temporários: prazo de inscrição entra na reta final
Seleção ocorre para a contratação imediata de até 300 vagas. Certame oferece vagas em seis áreas e os salários chegam a R$ 8,3 mil
PREVENÇÃO
Relembre as orientações do Ministério da Saúde no combate à dengue
Pasta reforça à população medidas essenciais de como se proteger, sobre diagnóstico e onde é possível se imunizar contra a doença
SAÚDE INDÍGENA
Malária: 230 profissionais qualificados para administrar tratamento inovador
Grande vantagem é que, enquanto o medicamento mais antigo precisa ser administrado ao longo de 14 dias, a tafenoquina deve ser tomada uma única vez
Acesso Rápido
Arboviroses






