




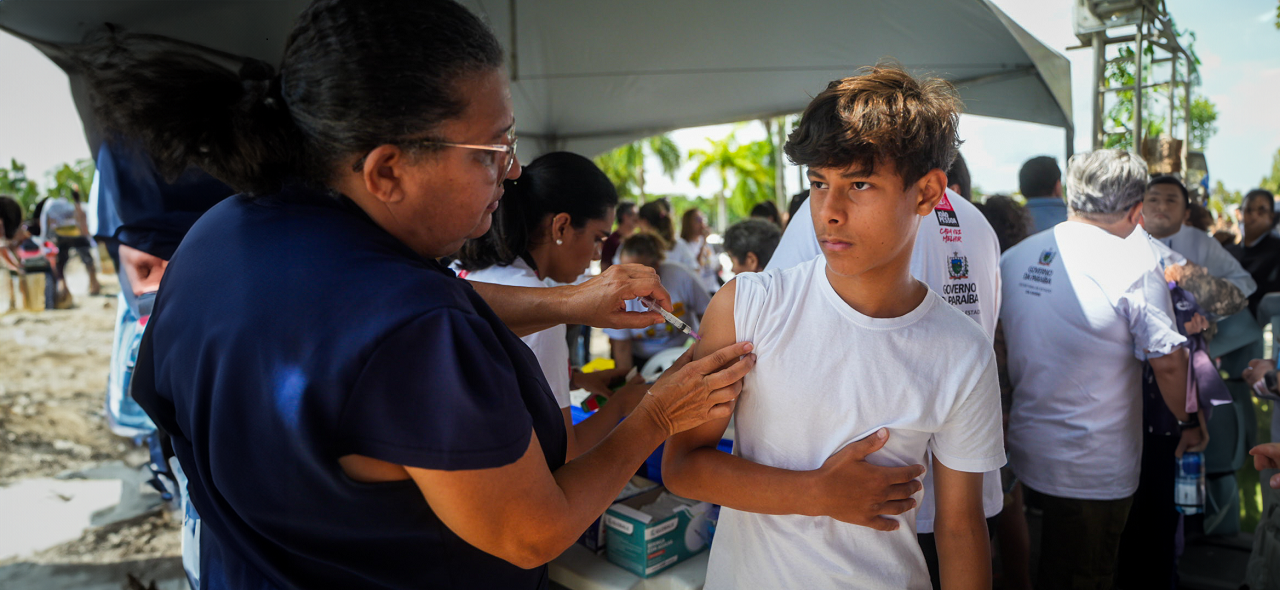
ESTRATÉGIA TEMPORÁRIA
Ministério da Saúde recomenda ampliação do público da vacina da dengue
Saiba mais sobre ESTRATÉGIA TEMPORÁRIA

ENFRENTAMENTO À DENGUE
Colóquio com diversos especialistas debate o atual cenário da dengue no país
Saiba mais sobre ENFRENTAMENTO À DENGUE

SAÚDE E INOVAÇÃO
Saúde e Google fazem parceria para facilitar acesso a informações sobre postos de vacinação
Saiba mais sobre SAÚDE E INOVAÇÃO

CONTRA O MOSQUITO
Em evento com especialistas, Saúde reforça tendência de queda nos casos de dengue
Saiba mais sobre CONTRA O MOSQUITO



NOVO PAC SAÚDE
Manual de orientações sobre nova etapa para gestores já está no ar
Estados e municípios têm até o dia 10 de maio para concluir a formalização das propostas selecionadas pelo Novo PAC
TELESSAÚDE
Proadi-SUS fortalece estratégia com investimento de R$ 133,6 milhões
Valores correspondem a recursos oriundos de imunidade tributária. Previsão é ofertar 160 mil atendimentos a usuários do SUS usando a saúde digital
SAÚDE INDÍGENA
Ministério da Saúde inicia plano para vacinar 130 mil indígenas
Ao longo do Mês de Vacinação dos Povos Indígenas, serão ofertadas 240 mil doses. Ação começou neste sábado (13)
Acesso Rápido
Arboviroses






